
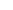 商标分类
商标分类  商标转让
商标转让 
一种去氧孕烯关键中间体的制备方法和去氧孕烯的制备方法与流程
 2021-02-02 21:02:34|
2021-02-02 21:02:34| 538|
538| 起点商标网
起点商标网
本发明属于甾体激素药物制备技术领域,具体是涉及到一种去氧孕烯关键中间体的制备方法和去氧孕烯的制备方法。
背景技术:
去氧孕烯是一种高效、低毒、无副作用的新一代甾体类避孕药,最早是由荷兰欧加农公司于1992年开发的口服避孕,具有广阔的市场前景。
去氧孕烯的合成路线较多,简要介绍如下:
1、us3927046公开了一种合成路线
化合物2可由左炔诺孕酮的中间体(沃氏物)经水解、发酵制备,来源方便,售价约50000-60000元/kg。
本法是经典的制备路线,无专利纠纷;方法成熟、收率尚可,国内较多企业采用此法制备。但此法用到恶臭的乙二硫醇,对研发、将来的生产均会产生极不利的影响;另外为提高收率,17-羟基先还原、再氧化,略嫌繁琐;文献未报道详细收率。
2、cn201210397815.9,公开了一条合成路线
此法先引入炔基,然后脱去3-保护,文献报道总收率37.6%,未见明显副反应,但此法乙二硫醇保护剂一直用到最后,且最后通过还原反应除去环状硫醚,故不可能消除成品中的异味,实际生产较难采用。
3、cn200810246594公开了合成方法
该法可以制备化合物8(z-13),本法简练,总收率38%,适合工业制备。但本法同样用到恶臭的乙二硫醇,必须考虑严格的劳动保护及废气的外漏,且17位用缩酮保护,产物非常不稳定,在一定温度或酸性条件下易分解。
技术实现要素:
本发明要解决的技术问题是提供一种去氧孕烯关键中间体的制备方法和去氧孕烯的制备方法,路线效率高,收率高,纯度更好。
本发明的内容为一种去氧孕烯关键中间体的制备方法,步骤为,将中间体iv溶于溶剂中,在还原剂硼氢化钠和路易斯酸的作用下,选择性还原17位酮基生成17位羟基,处理,得到中间体v,所述中间体iv和中间体v的结构式如下:
所述路易斯酸为氯化铈、三氯化铝或三氯化铁,优选为氯化铈,更优选7水氯化铈。
所述溶剂为二氯甲烷、四氢呋喃、甲醇、乙醇中的一种或多种。
所述溶剂为二氯甲烷和甲醇混合物,四氢呋喃和甲醇混合物或者四氢呋喃和乙醇混合物,最优选为四氢呋喃和甲醇混合物。
中间体iv和还原剂硼氢化钠的摩尔比为1:0.5~1:2.5,优选1:0.8;中间体iv和路易斯酸的质量比为1:0.8~1:2.0,优选1:1。
反应温度为-30~30℃,优选-10~15℃。
所述中间体iv的制备方法为,
将11-羟-乙基诺龙分散于甲醇中,在三氟化硼乙醚作用下,与乙二硫醇反应,反应温度为10-15℃,生成缩合物,水析过滤后,得到中间体ii;
中间体ii分散在溶剂(优选为四氢呋喃)中,和金属锂和液氨混合,反应完全后,经过淬灭,洗涤,析晶,得到中间体iii;
将中间体iii分散在溶剂(优选为丙酮或二氯甲烷)中,在-10~-15℃下,滴加琼斯试剂,反应完全后,经过淬灭,中和,水析得到中间体iv;
反应路线如下:
本发明提供一种去氧孕烯的制备方法,将中间体iv按照上述制备方法得到中间体v后,将中间体v溶解在甲苯和二甲基亚砜中,滴加到反应溶剂中,反应溶剂为甲基三苯基溴化磷、二甲基亚砜和氢化钠的混合物,反应完全后,淬灭,水洗,浓缩,析晶,过滤后,得到中间体vi;
将中间体vi分散在有机溶剂(优选为丙酮)中,加入2-碘酰基苯甲酸,反应完全后,过滤,浓缩,水析,得到中间体vii;
金属锂分散在乙二胺的溶液中,通炔气,得到反应体系,滴加中间体vii的四氢呋喃溶液,反应完全后,淬灭,浓缩,析晶得到去氧孕烯粗品;去氧孕烯粗品经过精炼,得到去氧孕烯;
反应路线如下:
去氧孕烯粗品精炼的步骤为,将去氧孕烯粗品分散在二氯甲烷溶剂中,加入活性炭脱色,过滤,浓缩,正己烷重析晶,得去氧孕烯精品。
中间体vi的制备方法中,反应溶剂的制备方法为,将甲基三苯基溴化磷和二甲基亚砜混合,搅拌下加入氢化钠,加热至75-85℃,维持一段时间得到。
去氧孕烯粗品的制备方法中,反应体系的制备方法为,在氮气保护下,加入乙二胺,持续通氮气,升温至55-65℃,改通炔气一段时间,加入金属锂,反应液全部变黑、温度升至100℃以上且反应液开始变白时,降温至80-85℃,反应液完全变白后保温反应,直至反应液面有油状物,变稀稠。
本发明的有益效果是,本发明提供了一种制备去氧孕烯中间体的新路线,且各个中间体稳定,不易分解。从11-羟基-乙基诺龙计算,到关键中间体v即3-去氧-11-酮基-17-羟基-乙基诺龙,此步重量收率可以达到70%以上,纯度98%以上;到成品去氧孕烯,该路线重量总收率达50%以上,纯度99%以上,符合药典要求
本方案采用以11-羟-乙基诺龙为起始物料,经双硫缩合、去氧、氧化、还原、witting、氧化、炔化,得到去氧孕烯.在17位酮基的保护反应步骤中,本方法采用更稳定的保护基团,得到的中间体iv更稳定,使整条路线效率高,收率高,纯度更好。
本发明将17位选择性还原作为羟基的中间体结构比较稳定,现有技术一般采用17位作为缩酮保护,其底物极其不稳定,不耐酸,耐温,30℃左右就会变坏;选择性还原的稳定性更好,收率和纯度更好。
附图说明
图1为本发明的反应路线图。
具体实施方式
实施例1制备中间体ii
在三口瓶中向加入甲醇120g,投入11-羟基乙基诺龙30g,调温10~15℃,加入乙二硫醇12g,搅拌10分钟。控制温度10~15℃,向反应罐中滴加三氟化硼乙醚溶液9ml(滴加时放热明显),于10~15℃保温反应1小时15分钟,取样进行tlc检测至原料点消失.反应完全后,将反应液倒入420ml冰水中水析,降温0~5℃,搅拌30分钟,过滤,滤饼水洗至中性,甩干,出料,将物料放入40~50℃烘干12~16小时,得中间体ii。收率:100~110%。
实施例2制备关键中间体iii
三口瓶ii中,降温至-70℃,通液氨,待通液氨至500ml时,停止通氨气。称取6.5g金属锂,每次间隔10分钟,分三次加入反应瓶中,控制内温不高于-50℃,加锂完毕后,保温搅拌2小时。向反应瓶中滴加中间体ii50g的四氢呋喃溶液250ml,内温保持在-38.0℃以下,滴加时间约0.5小时。滴加完毕后,继续保温搅拌反应1.5小时开始点板,tlc检测原料点消失。反应结束后,向反应瓶内缓慢滴加无水乙醇80g,滴加时控制内温在-38.0℃以下,滴加完毕继续搅拌20分钟,停止降温。缓慢升温至室温,做好尾气吸收,室温下搅拌2小时,温度控制在25.0~30.0℃,真空度控制在-0.06mpa以下,浓缩四氢呋喃至无液体滴出.浓缩完毕,滴加10%naoh溶液,滴加过程中罐内温度不高于30.0℃,滴加完搅拌1.5小时,当罐内温度降至25.0℃时,进行水析,水析完毕后,继续搅拌约4小时。滤饼用饮用水水洗,水洗至中性,物料放入40~50℃烘干10~16小时,得中间体iii。收率:70~80%。
实施例3制备中间体iv
三口瓶中加入350g丙酮和20g地-去酮物,降温至-15.0℃时,开始滴加19.6ml琼斯试剂,滴加过程中温度保持在-10.0~-15.0℃,滴加时间约2小时。滴加结束后取样点板至原料点消失,空温在-10.0~-15.0℃下加入20g异丙醇,时间约10分钟,搅拌10分钟。加入碳酸钠饱和溶液98ml直至溶液呈微碱性,将反应液倒入600ml冰水中水析,滤饼水洗至中性,甩干,物料在温度30.0~35.0℃下,干燥20小时,得中间体iv,收率80-90%。纯度≥95.0%。
实施例4制备中间体v
三口瓶中,加入中间体iv20g,二氯甲烷100ml,甲醇ml,cecl3·7h2o20g,室温下搅拌溶解,待全部溶解后,降温至0℃。分四次加入硼氢化钠,每次间隔5min,加毕,继续在0℃下反应。tlc监控反应完全,冰醋酸和水加入至三口瓶中,45℃下浓缩有机相至体系有固体生成,降温至0-5℃左右,冷冻1-2h,过滤,5v水淋洗,白色固体加入4v甲苯,搅拌,在50℃下减压浓缩至干,得到中间体v,用卡费希尔法检测水分,使其水分小于0.1,(若大于0.1,继续加入甲苯,浓缩带水),收率80-85%,hplc大于98%。
实施例5制备中间体vi
氮气保护下,三口瓶中,加入甲基三苯基溴化磷15g,dmso100ml,搅拌下加入nah3.0g,加入nah后放热明显,待反应体系稳定后,搅拌下加热至80度左右,维持至少2h;而后滴加中间体v10g的dmso/甲苯溶液,滴加完后,保温80℃左右反应,tlc监测,大约2-3h反应完毕,而后搅拌下将反应液倒入30v中,分出有机相,水相用甲苯萃取3次(3v*3),合并有机相,水洗2次(5v*2),45℃减压浓缩至干,加入8v乙醇,减压浓缩至干,加入8v乙醇,搅拌下滴加30v水,而后45℃减压浓缩除去丙酮降温至0℃析晶至少1h,过滤,水洗2次,湿品加入单口瓶中,加入10v乙醇,加热待溶清后,降温至20℃,滴加5v左右的水,过滤,冷丙酮:水=2:1洗涤,50℃干燥。纯度99%,收率75-80%。
实施例6制备中间体vii
三口瓶中,加入中间体vi10g,丙酮50ml,dmso50ml,搅拌下溶解,再加入ibx(2-碘酰基苯甲酸)20g,tlc监控反应完全,过滤,5v丙酮淋洗滤饼,减压浓缩至无溶剂滴出,加入50ml10%的硫代硫酸钠溶液,再加入20v水,搅拌1h,过滤,滤饼用水洗涤,50℃干燥。收率85%-90%。纯度大于98%。
实施例7制备去氧孕烯
三口瓶中,氮气保护下,加入乙二胺100g,持续(液底)通氮气,15分钟后开始升温至60℃改通炔气,25分钟,加入金属锂5g,(观察温度变化)。温度自然上升,反应液全部变黑,温度升至100℃以上时,观察反应液开始变白,降温85-80℃,反应液完全变白后保温反应1小时。反应液面有油状物,变稀稠状。(如反应液没变稠继续延长反应时间)。主料配制(10g中间体vii,30g四氢呋喃),反应液降温至20~25℃,开始滴加(约滴加20-25分钟,滴加完后,保温反应1.5小时,点板至原料点变浅粉色。水析罐加入100g水,30g氯化铵,降温-15℃以下,将反应液缓慢滴入氯化铵水溶液中,分出水层,水层用正己烷150g提取5次,每次用30g,搅拌2分钟,静止10分钟,合并有机层,水洗至中性。加入无水硫酸镁50g,搅拌脱水1小时,常温搅拌40分钟,过滤至浓缩罐中,滤饼用20g正庚烷淋洗一并抽入浓缩罐中水浴减压浓缩近干,加入正庚烷20g,升温溶清后放入不锈钢桶中,至大量结晶析出,冷却30分钟。甩滤,滤饼用约0.5l正庚烷淋洗,甩干,30℃干燥。收率85%-90%。纯度大于98%。
实施例8制备去氧孕烯的精制
三口瓶中,加入70g二氯甲烷,10g去氧孕烯粗品,搅拌使其全溶,再加入活性炭,搅拌1-1.5h,过滤,减压浓缩至干。将20ml正己烷/异丙醚的混合溶媒加入结晶罐中,升温至55~60℃,搅拌使物料溶解,搅拌下降温至0℃左右。析晶1-2h,离心,用约0.5l正己烷淋洗滤饼,甩干.摊盘,45~50℃干燥7小时,得去氧孕烯精品,收率,80%,纯度大于99%,符合药典要求。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



