
 商标分类
商标分类  商标转让
商标转让 
一种吉西他滨中间体的高选择性的合成方法与流程
 2021-02-02 21:02:56|
2021-02-02 21:02:56| 572|
572| 起点商标网
起点商标网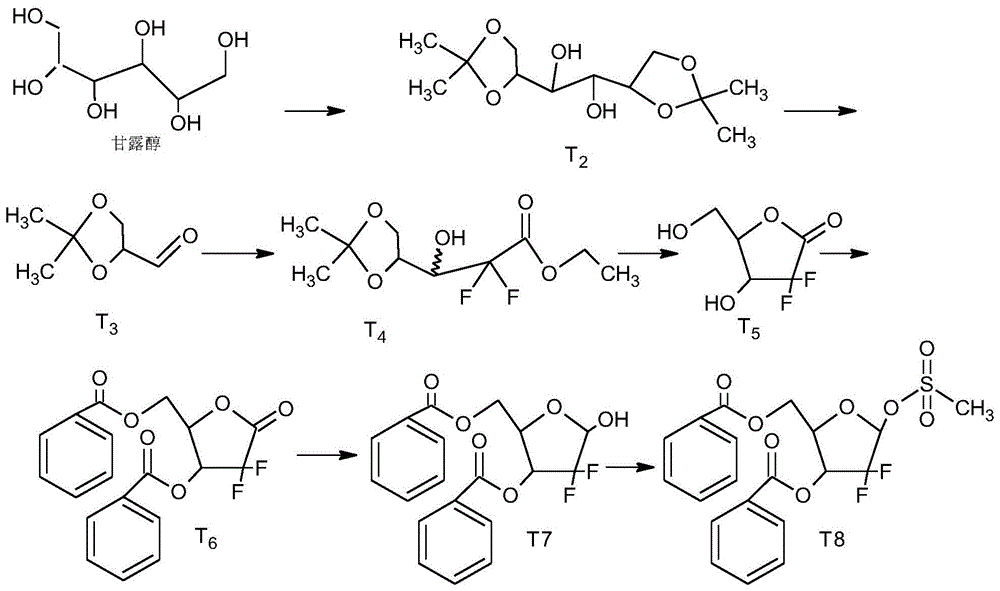
本发明涉及药物合成化学领域,具体涉及吉西他滨中间体的高选择性的合成方法。
背景技术:
盐酸吉西他滨是一种人工合成的新型二氟核苷类抗代谢抗肿瘤药,该品首先由美国elililly公司开发研制,商品名“健择"。盐酸吉西他滨可抑制dna的合成;作为前药,盐酸吉西他滨被细胞摄入后,经脱氧胞苷激酶磷酸化吉西他滨单磷酸酯,进一步转变为活性代谢物吉西他滨二磷酸酯或三磷酸酯。吉西他滨三磷酸酯可竞争性抑制dna链的延长,导致dna片段形成和细胞死亡,这种抑制活性可通过吉西他滨二磷酸酯和吉西他滨三磷酸酯的自身强化性质得到增强。其结果是在细胞内维持高浓度的吉西他滨代谢物,降低其竞争性抑制物脱氧胞苷三磷酸酯的细胞内浓度。吉西他滨磷酸化激活的效率比ara-c强6倍。吉西他滨还具有独特的自我增强机制,可能包括代谢活化的产物对核苷激酶有更高的亲和力、穿透靶细胞膜能力的增加、药物排泄的减缓,从而使活化的核苷在细胞内持续高水平积聚,这一特点对于抗肿瘤效应的提高也很有意义。盐酸吉西他滨的独特药理机制和潜在价值带来了巨大的市场潜力,自上市以来,己经成为局限晚期或转移性非小细胞肺癌的一线药物,是全球市场上的十大抗肿瘤药之一。1995年在南非、瑞典、荷兰、澳大利亚等国家获准上市,1996年美国fda也已批准,临床用于治疗非小细胞肺癌和胰腺癌的一线治疗。1999年经我国sfda批准治疗非小细胞肺癌的一线用药。目前盐酸吉西他滨的临床的适应症有非小细胞肺癌、胰腺癌、膀肤癌、乳腺癌及其他实体肿瘤。
上式为吉西他滨盐酸盐的分子式,吉西他滨盐酸盐是一种新型的人工合成的细胞周期性特异性抗代谢类抗肿瘤药物,属于核苷类似物,化学名称为2-脱氧-2,2-二氟-b-核胞苷盐酸盐,其合成的关键中间体是2-脱氧-2,2-二氟-d-赤式-戊呋喃糖-3,5-二苯甲酯-1-甲磺酸酯(t8),以d-甘露醇为起始原料,用乙二醇二甲醚为溶剂,在无水氯化亚锡催化下,2,2-二甲基丙烷与1,2和5,6位羟基选择性反应得到t2,t2在二氯甲烷溶剂里面用高碘酸钠氧化,得到甘油醛缩丙酮t3,甘油醛缩丙酮t3和二氟溴乙酸乙酯发生reformatsky反应得到t4,t4在三氟乙酸催化下脱保护并关环得到t5,t5在dmap催化下和苯甲酰氯酯化反应得到t6,t6在三叔丁基氢化铝锂还原下反应成t7,t7再和甲基磺酰氯反应得到t8,见附图1。
由上述合同路线合成的吉西他滨盐酸盐的过程中,存在以下四点问题:
(1)、t6→t7的合成过程中需在超低温(-60℃以下)下反应,这样就需要消耗大量的能源及增加深冷设备,如液氮制冷装置。
(2)、t6→t7的合成过程中使用的还原剂三叔丁基氢化铝锂不稳定,需要自己制备,用氢化铝锂和叔丁醇在乙醚溶剂下反应,氢化铝锂遇水非常容易燃烧,乙醚也易着火,生产上存在较大的安全风险。
(3)、t7→t8合成过程中,产品t8的所需要的异构体a含量低,需要结晶得到高含量的异构体a使用,结晶母液中含有20%左右产品无法利用,而作为副产物废去,这样不仅造成收率低下,成本高而且对环境污染大,增加三废的处理成本。
技术实现要素:
为了达到上述目的,本发明提出了本发明旨在提供一种经济效益高效益高的吉西他滨中间体的高选择性的合成方法,具体技术方案如下:
一种吉西他滨中间体的高选择性的合成方法,具体包括如下工艺:
step1,t1的合成:
向反应釜中加入d-异抗坏血酸、5,6位的羟基保护试剂、丙酮溶剂、反应催化剂,控制反应完全,加入碳酸钠水溶液淬灭反应,水相含产品t1;
step2,t2的合成:
将t1中滴加550kg双氧水,控制反应生成t2;
step3,t3的合成:
向反应釜中加三水乙酸钠或者碳酸钠,用冰醋酸调节ph值,滴加10-15%次氯酸钠水溶液,控制反应生成t3;
step4,t4的合成:
向干燥反应釜中加四氢呋喃、二氟溴代乙酸乙酯和t3搅拌抽入高位槽,在反应釜中加入四氢呋喃、锌粉及活化剂,滴加反应,控制在60~70℃之间反应2小时,降温,加入乙酸乙酯;滴加5%盐酸控制ph=3~4,控制温度15℃以下,分层,有机相依次用碳酸氢钠,饱和盐水洗涤,无水硫酸钠干燥等后处理后,减压将溶剂蒸干,得产品t4;
step5,t5的合成:
向反应釜中加入t4,水,盐酸,混合搅拌升温至70-80℃反应,水相减压蒸干,加入乙腈常压蒸馏带水直到关环完全后减压蒸干,得到产品t5;
step6,t6的合成:
向反应釜中加入有机溶剂溶解t5、吡啶、dmap,升温至30~50℃,缓慢滴加苯甲酰氯,滴加完毕后,反应8小时后加入无水硫酸钠和活性炭,氮气保护,过滤,滤饼用有机溶剂洗涤,温度控制在80℃下减压蒸馏回收有机溶剂,至无馏分;降温加入二氯甲烷和石油醚,30~40℃搅拌溶解,降温至0~5℃搅拌结晶2小时,氮气保护下离心甩滤,冷石油醚淋洗,烘干得产品t6;
step7,t7的合成:
向反应釜中抽入无水四氢呋喃,并通氮气保护,称取t6投入反应釜,搅拌溶解,向高位槽加入无水四氢呋喃,再加入红铝50kg,并通氮气保护,搅拌下向反应釜中滴加红铝溶液,滴加完毕后再向高位槽中再加四氢呋喃淋洗;反应2小时之后关闭氮气,冷却搅拌下加入二氯甲烷,并缓慢加入盐酸,并静止分层;再对有机相先用5%的小苏打水洗一次,再用饱和盐水洗一次,静置分层后除去水相,向反应釜加入无水硫酸钠进行干燥,之后进行过滤,并在-0.09mpa、50℃以下浓干溶剂,得到黄色透明粘液t7;
step8,t8的合成:
向反应釜中加入二氯甲烷,加入t7并搅拌溶解,之后通氮气保护再通盐水降温,将温度控制在-5℃以下后向反应釜中抽入三乙胺,将温度控制在-5度以下,在高位槽抽入甲烷磺酸氯,再抽入二氯甲烷,搅拌下向反应釜慢慢滴加,温度控制在-10-5℃,反应完全后,关闭氮气并在搅拌下向反应釜中滴加盐酸并将温度控制在0度以下,之后搅拌分层,将有机层再用碳酸氢钠洗涤,调节ph至中性,再向反应釜加入无水硫酸钠并干燥,之后过滤减压浓缩滤液,保持压力为-0.09mpa、温度为50℃以下,得到黄色透明粘液t8;
step9,t8构型转化:
向反应釜中加入甲苯、三乙胺、甲基磺酸,将其缓慢加热至回流,并保持回流6h,之后将其冷却至常温后用10%盐酸溶液洗涤分层,之后收集有机层,再将有机层再用5%小苏打溶液洗涤,之后加入元明粉干燥2小时后过滤,之后将其减压浓缩干溶剂得到淡红色粘液,之后将其冷却至50-60℃并加入乙醇,再将盐水冷却至-2℃并结晶5小时,将离心机甩干得到产品,母液浓干后重复t8构型转化工艺,再次得到产品;
所述b2化学名称:1,2:5,6-甘露醇二缩酮;b3化学名称:d-(r)-甘油醛缩丙酮;b4化学名称:(3r,s)-2,2-二氟-3-羟基-(2,2-二甲基二氧环戊-4-基)丙酸乙酯;t1化学名称;5,6-o-异亚丙基-d-异抗坏血酸;t2化学名称:3,4-o-异亚丙基-2-羟基-丁酸;t3化学名称:2,3-o-异亚丙基-甘油醛;t4化学名称:(3r,s)-2,2-二氟-3-羟基-(2,2-二甲基二氧环戊-4-基)丙酸乙酯;t5化学名称:2-脱氧-2,2-二氟-1-羰基核糖;t6化学名称:2-脱氧-2,2-二氟-d-赤型-1-呋喃酮糖-3,5-二苯甲酰酯。
进一步的,步骤step1所述羟基保护催化剂为对甲苯磺酸、浓硫酸、浓盐酸等质子酸和能够使反应产生酸性环境的路易斯酸中的一种,如无水氯化亚锡,氯化锌等。
进一步的,步骤step2采用氧化剂为双氧水和次氯酸钠中的一种,该氧化剂在水中反应,反应过程条件温和,容易控制,副产物少,成本很低。
进一步的,步骤step3次氯酸钠可用氯气等氧化剂代替。
进一步的,步骤step7向反应釜中抽入无水四氢呋喃,并通氮气保护时,将温度控制在-5℃以下。
进一步的,步骤step7向高位槽加入无水四氢呋喃,再加入红铝50kg,并通氮气保护,搅拌下向反应釜中滴加红铝溶液,将温度控制在-5℃以下。
进一步的,步骤step7中反应2小时之后关闭氮气,冷却搅拌下加入二氯甲烷,并缓慢加入质量分数为10%的盐酸,同时保持温度在0-5℃并静止分层。
进一步的,步骤step8中将有机层再用5%碳酸氢钠洗涤,调节ph至中性,再向反应釜加入无水硫酸钠并干燥2小时。
由于上述技术方案的实施,本发明与现有技术相比具有下列优点:
一、本发明利用sn2反应,在t8反应体系中,加入甲苯作溶剂,反应试剂甲基黄酸和三乙胺,将异构体比例由初始的1.5:1提高到3.5:1,通过此步骤,可以将产品的收率提高10%,成本可以有效的降低13万/吨。
二、本发明将结晶母液加入甲苯作溶剂,反应试剂甲基黄酸和三乙胺,再度进行构型转换,提炼出需要的a异构体,通过此步骤,可以将产品的收率提高5%,成本可以有效的降低7.5万/吨。
三、本发明不使用超低温反应后,每吨产品将减少能源费3万/吨,通过这3项技术创新后,总计可以将成本有效降低23.5万/吨,如果每年销售50吨,将给企业取得直接经济效益1175万元/年。
附图说明
图1为吉西他滨中间体的传统合成路线图。
图2为吉西他滨中间体的高选择性的合成方法中盐酸吉西他滨中间体的合成路线一的合成路线图。
图3为吉西他滨中间体的高选择性的合成方法中盐酸吉西他滨中间体的合成路线二的合成路线图。
具体实施方式
下面将结合具体的实施例和附图对本发明做出进一步的描述。
具体技术方案如下:
一种吉西他滨中间体的高选择性的合成方法,具体包括如下步骤:
step1,t1的合成:
向反应釜中加入d-异抗坏血酸、5,6位的羟基保护试剂、丙酮溶剂、反应催化剂,控制反应完全,加入碳酸钠水溶液淬灭反应,水相含产品t1;
step2,t2的合成:
将t1中滴加550kg双氧水,控制反应生成t2;
step3,t3的合成:
向反应釜中加三水乙酸钠或者碳酸钠,用冰醋酸调节ph值,滴加10-15%次氯酸钠水溶液,控制反应生成t3;
step4,t4的合成:
向干燥反应釜中加四氢呋喃、二氟溴代乙酸乙酯和t3搅拌抽入高位槽,在反应釜中加入四氢呋喃、锌粉及活化剂,滴加反应,控制在60~70℃之间反应2小时,降温,加入乙酸乙酯;滴加5%盐酸控制ph=3~4,控制温度15℃以下,分层,有机相依次用碳酸氢钠,饱和盐水洗涤,无水硫酸钠干燥等后处理后,减压将溶剂蒸干,得产品t4;
step5,t5的合成:
向反应釜中加入t4,水,盐酸,混合搅拌升温至70-80℃反应,水相减压蒸干,加入乙腈常压蒸馏带水直到关环完全后减压蒸干,得到产品t5;
step6,t6的合成:
向反应釜中加入有机溶剂溶解t5、吡啶、dmap,升温至30~50℃,缓慢滴加苯甲酰氯,滴加完毕后,反应8小时后加入无水硫酸钠和活性炭,氮气保护,过滤,滤饼用有机溶剂洗涤,温度控制在80℃下减压蒸馏回收有机溶剂,至无馏分;降温加入二氯甲烷和石油醚,30~40℃搅拌溶解,降温至0~5℃搅拌结晶2小时,氮气保护下离心甩滤,冷石油醚淋洗,烘干得产品t6;
step7,t7的合成:
向1000l反应釜中抽入250kg无水四氢呋喃,并通n2保护,温度控制在-5--10度以下,准确称50kgt6投入反应釜,搅拌溶解。向高位槽加入无水四氢呋喃100kg,再加入红铝50kg,并通n2保护,搅拌下向反应釜中滴加红铝溶液,温度控制在-5度以下。滴加毕,高位槽中再加少量四氢呋喃淋洗,加入反应釜中,反应2小时至反应完全。关闭n2,冷却搅拌下,加入300kg二氯甲烷,缓慢加入250kg10%的盐酸,保持温度在0-5度,分层。有机相用5%的小苏打水洗一次,有机层再用100kg饱和盐水洗一次,静置分层,分去水相。向反应釜加入25kg无水硫酸钠,干燥2小时。过滤,-0.09mpa,50℃以下浓干溶剂,得到黄色透明粘液t7,称重得到55kg产品;
step8,t8的合成:
向1000l反应釜中加入250kg二氯甲烷,加55kgt7,搅拌溶解,通n2保护再通盐水降温,温度控制在-5度以下。向反应釜中抽入三乙胺25kg,温度控制在-5度以下。高位槽抽入甲烷磺酸氯20kg,再抽入100kg二氯甲烷,搅拌下向1000l反应釜慢慢滴加,温度控制在-10—-5度。1小时内滴加完,反应完全后,关闭n2,搅拌下向反应釜中滴加1n盐酸100kg,温度控制在0度以下。搅拌,分层,有机层再用5%碳酸氢钠200kg洗涤,调节ph=7左右。向反应釜加入25kg无水硫酸钠,干燥2小时。过滤.减压浓缩滤液,-0.09mpa,50℃以下,得到黄色透明粘液t8;
step9,t8构型转化:
向500l反应釜中加入200kg甲苯,20kg三乙胺,10kg甲基磺酸。缓慢加热至回流,保持回流6h。冷却至常温用10%盐酸溶液洗涤分层,收集有机层。有机层再用60kg5%小苏打溶液洗涤。加入25kg元明粉干燥2小时,过滤。减压浓缩干溶剂得到淡红色粘液。冷却至50-60度,加入400kg乙醇。盐水冷却至零下2度结晶5小时,离心机甩干得到产品51kg,母液浓干后重复t8构型转化工艺,再次得到产品2.5kg;
所述b2化学名称:1,2:5,6-甘露醇二缩酮;b3化学名称:d-(r)-甘油醛缩丙酮;b4化学名称:(3r,s)-2,2-二氟-3-羟基-(2,2-二甲基二氧环戊-4-基)丙酸乙酯;t1化学名称;5,6-o-异亚丙基-d-异抗坏血酸;t2化学名称:3,4-o-异亚丙基-2-羟基-丁酸;t3化学名称:2,3-o-异亚丙基-甘油醛;t4化学名称:(3r,s)-2,2-二氟-3-羟基-(2,2-二甲基二氧环戊-4-基)丙酸乙酯;t5化学名称:2-脱氧-2,2-二氟-1-羰基核糖;t6化学名称:2-脱氧-2,2-二氟-d-赤型-1-呋喃酮糖-3,5-二苯甲酰酯。
其中,步骤step1所述羟基保护催化剂为对甲苯磺酸、浓硫酸、浓盐酸等质子酸和能够使反应产生酸性环境的路易斯酸中的一种,如无水氯化亚锡,氯化锌等。
其中,步骤step2采用氧化剂为双氧水和次氯酸钠中的一种,该氧化剂在水中反应,反应过程条件温和,容易控制,副产物少,成本很低。
其中,步骤step3次氯酸钠可用氯气等氧化剂代替。
其中,步骤step7向反应釜中抽入无水四氢呋喃,并通氮气保护时,将温度控制在-5℃以下。
其中,步骤step7向高位槽加入无水四氢呋喃,再加入红铝50kg,并通氮气保护,搅拌下向反应釜中滴加红铝溶液,将温度控制在-5℃以下。
其中,步骤step7中反应2小时之后关闭氮气,冷却搅拌下加入二氯甲烷,并缓慢加入质量分数为10%的盐酸,同时保持温度在0-5℃并静止分层。
其中,步骤step8中将有机层再用5%碳酸氢钠洗涤,调节ph至中性,再向反应釜加入无水硫酸钠并干燥2小时。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



