
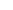 商标分类
商标分类  商标转让
商标转让 
N-烷氧基受阻胺光稳定剂及其中间体的制备方法与流程
 2021-02-02 13:02:45|
2021-02-02 13:02:45| 558|
558| 起点商标网
起点商标网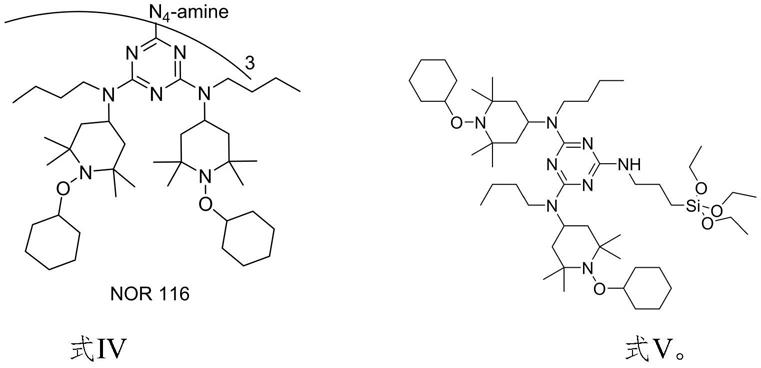
n-烷氧基受阻胺光稳定剂及其中间体的制备方法
技术领域
[0001]
本发明涉及化学合成领域,尤其涉及n-烷氧基受阻胺光稳定剂及其中间体的合成方法。
背景技术:
[0002]
传统的hals光稳定剂在哌啶环上存在n-h基团,具有一定碱性,使其在酸性树脂、酸性配合剂和酸性环境下的应用受到限制,为拓宽其应用领域,低碱性化研究成为hals光稳定剂的研究热点,尤其n-烷氧基受阻胺,已有nor116、tinuvin123、tinuvin 152、nor371、la-81等产品上市,其中作为市面中第一款具备光稳定性能和一定阻燃性的稳定剂,nor 116的面世推动了无卤化阻燃剂的发展。n-烷氧化受阻胺基三嗪类中间体(2-氯-4,6-双[n-(1-环己氧基-2,2,6,6-四甲基哌啶-4-基)丁基氨基]三嗪)作为nor 116制备过程中的一种十分重要的中间体,其高效的制备能有效推动低碱性受阻胺光稳定剂的研发工作。
[0003]
专利ep2035381a公开了使用醛类作烷基化试剂,使用th-7作为起始原料,然后采用两步法制备nor116中间体:首先利用过氧乙酸催化2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪得到双氮氧自由基化合物,对双氮氧自由基化合物后处理之后烷基化得到nor 116中间体,即2-氯-4,6-双[n-(1-环己氧基-1,2,2,6,6-四甲基哌啶-4-基)丁基氨基]三嗪。该工艺路线存在后处理工艺复杂的问题,多次清洗后采用柱层析的方法进行的层析;同时反应效率较低,第一步收率70%,第二步投入1g原料进行环己基化,使用柱层析得到100mg目标产物,收率仅为8%。
技术实现要素:
[0004]
鉴于以上问题,我们需要提出一种制备过程简单高效,后处理工艺便捷的方法来得到我们目标的中间体产物。
[0005]
本发明的目的是提供一种n-烷氧基受阻胺光稳定剂中间体的制备方法,采用一锅法,以式i所示的2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪为原料,与过氧化物发生氧化反应生成式ii所示的自由基中间体,式ii所示的自由基中间体与环己基甲醛发生烷基化反应,制得式iii所示的目标产物;
[0006][0007]
本发明中,所述制备方法在乙酸酯类溶剂或c
1-c
8
的醇类溶剂条件下反应;
[0008]
优选地,所述乙酸酯类溶剂选自乙酸乙酯、乙酸丁酯、乙酸异丙酯、乙酸甲酯、乙酸
异丁酯、乙酸异戊酯中的一种或多种;所述c
1-c
8
的醇类溶剂选自甲醇、乙醇、异丙醇中的一种或多种。
[0009]
本发明中,所述烷基化反应在金属催化剂作用下进行;所述金属催化剂选自cu
2+
、cu
+
、fe
2+
、fe
3+
、co
2+
、mg
2+
的盐或氢氧化物;
[0010]
优选地,所述金属催化剂为mg(oh)
2
、mgcl
2
、cucl、cuoac、cui、cu
2
so
4
、cu(oac)
2
、cuso
4
、cuco
3
、cucl
2
、feso
4
、fecl
2
、cocl
2
中的一种或多种。
[0011]
本发明中,金属催化剂的用量为2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪的摩尔量的2-8mol%。
[0012]
本发明中,
[0013]
当以乙醇为反应溶剂时,金属催化剂选择cu
+
盐或氢氧化物;
[0014]
当以甲醇或乙酸酯类溶剂为反应溶剂时,金属催化剂选择cu
2+
盐或氢氧化物;
[0015]
当以异丙醇为反应溶剂时,金属催化剂选择cu
2+
盐或mg
2+
盐。
[0016]
本发明中,所述过氧化物选自过氧化氢、过氧乙酸、叔丁基过氧化氢中的一种或多种;
[0017]
优选地,过氧化物与2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪的摩尔比为4-10:1。
[0018]
本发明中,所述氧化反应在助剂作用下进行,所述助剂为过渡金属盐、镁盐或氧化物或氢氧化物;优选地,所述过渡金属盐优选为铜盐或铁盐,更优选为氯化亚铁、硫酸铜、氯化铜、碳酸铜中的一种或多种;所述镁盐或氧化物或氢氧化物选自氯化镁、硫酸镁、氢氧化镁、氧化镁中的一种或多种。
[0019]
本发明中,所述反应的反应温度为10-80℃,优选为40-65℃。
[0020]
本发明中,所述反应还包括采用结晶法提纯式iii所示的目标产物的步骤。
[0021]
本发明还提供n-烷氧基化的受阻胺光稳定剂的制备方法,上述方法制得式iii所示的n-烷氧基受阻胺光稳定剂中间体,式iii所示的n-烷氧基受阻胺光稳定剂中间体与三乙四胺或硅烷偶联剂kh550反应,制得如下式iv和式v所示的n-烷氧基化的受阻胺光稳定剂:
[0022][0023]
本发明取得了如下积极效果:采用一锅法制备n-烷氧基受阻胺光稳定剂中间体,不需要对自由基中间体提纯,该工艺得到的目标产物收率高、纯度好、反应时间短、后处理简单;操作方便。
具体实施方式
[0024]
以下结合具体实施方式详述本发明,但需说明的是,本发明的保护范围不受这些具体实施方式和原理性解释的限制,而是由权利要求书来确定。
[0025]
本发明中,除了明确说明的内容之外,未提到的任何事宜或事项均直接适用本领域已知的那些而无需进行任何改变。而且,本文描述的任何实施方式均可以与本文描述的一种或多种其他实施方式自由结合,由此形成的技术方案或技术思想均视为本发明原始公开或原始记载的一部分,而不应被视为是本文未曾披露或预期过的新内容,除非本领域技术人员认为该结合明显不合理。
[0026]
本发明所公开的所有特征可以任意组合,这些组合应被理解为本发明所公开或记载的内容,除非本领域技术人员认为该组合明显不合理。
[0027]
本说明书所公开的数值点,不仅包括实施例中具体公开的数值点,还包括说明书中各数值范围的端点,这些数值点所任意组合的范围都应被视为本发明已公开或记载的范围。
[0028]
本发明中的技术和科学术语,给出定义的以其定义为准,未给出定义的则按本领域的通常含义理解。
[0029]
一种n-烷氧基受阻胺光稳定剂中间体的制备方法,采用一锅法,以式i所示的2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪为原料,与过氧化物发生氧化反应生成式ii所示的自由基中间体,式ii所示的自由基中间体与环己基甲醛发生烷基化反应,制得式iii所示的目标产物;
[0030][0031]
本发明在自由基中间体产生的同时加入能与其发生烷基化反应的烷基化试剂环己基甲醛,使烷基化反应与氧化反应同时进行,制得目标产物。在具体实施例中,可以在氧化反应起始就加入烷基化试剂,也可以让氧化反应制得的自由基中间体有一定积累(如反应1-6h)后再加入环己基甲醛进行烷基化反应。该工艺通过烷基化反应不断地消耗氧化反应产生的自由基中间体,使得氧化反应能不断地正向进行,提高了反应的效率,而且发明人意外发现,氧化反应和烷基化反应同时进行还减少了副产物的产生,提高了目标产物的选择性。因此本发明的工艺不但提高了目标产物的收率,还减少了副产物的产生、提高了目标产物的选择性,也缩短了反应时间,节约成本。
[0032]
研究发现,所述氧化反应和烷基化反应在乙酸酯类溶剂或c
1-c
8
的醇类溶剂条件下进行,反应物与催化剂均有较好的相容性,有利于反应的进行和提高反应效率。本发明所述的“乙酸酯类溶剂”是指由乙酸与c
1-8
醇反应失水而生成的化合物,如乙酸乙酯、乙酸丁酯、乙酸异丙酯、乙酸甲酯、乙酸异丁酯、乙酸异戊酯。所述“c
1-c
8
的醇类溶剂”是指分子中含有与c
1-c
8
烃基侧链上的碳结合的羟基的化合物,如甲醇、乙醇、乙二醇、异丙醇、丁醇、戊醇、己
醇、庚醇、辛醇。
[0033]
本发明中所述乙酸酯类溶剂优选乙酸乙酯、乙酸丁酯、乙酸异丙酯、乙酸甲酯、乙酸异丁酯、乙酸异戊酯中的一种或几种;所述醇类溶剂优选甲醇、乙醇、异丙醇中的一种或几种。此种溶剂环境下,反应效率高,目标产物选择性好。所述溶剂与2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪的体积质量比(ml/g)为2-20:1,优选8-15:1;控制溶剂加入量在该范围内,可以使整个反应体系的相容性更佳,获得更高的产品收率。
[0034]
所述烷基化反应在金属催化剂作用下进行,所述金属催化剂选自cu
2+
、cu
+
、fe
2+
、fe
3+
、co
2+
、mg
2+
的盐或氢氧化物,优选地,所述金属催化剂为mg(oh)
2
、mgcl
2
、cucl、cuoac、cui、cu
2
so
4
、cu(oac)
2
、cuso
4
、cuco
3
、cucl
2
、feso
4
、fecl
2
、cocl
2
中的一种或多种。控制金属催化剂的加入量为2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪的摩尔量的2-8mol%,烷基化反应更彻底,在具体实施例中可以为3mol%、4mol%、5mol%、6mol%、7mol%。
[0035]
研究发现,以上反应溶剂和金属催化剂任意配合使用,都能使得反应的收率达到60%以上。进一步研究发现,当选用以下几种反应溶剂和金属催化剂配合使用时,收率能够进一步提高到90%左右。
[0036]
当以乙醇为反应溶剂时,烷基化反应的金属催化剂选择cu
+
盐或氢氧化物;尤其选用氯化亚铜、醋酸亚铜、碘化亚铜、硫酸亚铜,反应体系相容性好,可以获得较高的收率。
[0037]
当以甲醇或乙酸酯类溶剂为反应溶剂时,烷基化反应的金属催化剂选择cu
2+
盐或氢氧化物,尤其选用醋酸铜或碳酸铜作为烷氧基化反应催化剂具有较佳的催化效果。
[0038]
当以异丙醇为反应溶剂时,烷基化反应的金属催化剂选择cu
2+
盐或mg
2+
盐,尤其选用醋酸铜、氯化镁,反应体系相容性好,可以获得较高的收率。
[0039]
研究发现,在甲醇或乙酸酯类溶剂体系中如果选用cu
+
盐,比如使用氯化亚铜作为催化剂时,反应体系中会形成胶状物,该胶状物粘在反应设备和搅拌装置上,影响反应的进行,进一步影响催化效果,反应收率低。
[0040]
研究发现,以间二甲苯为溶剂时,该溶剂体系与金属催化剂的相容性较差,特别地,与cucl、cu(oac)
2
的相容性最差,催化剂与溶剂为固液两相体系。此外,在此溶剂体系下,氧化反应效率极低,尤其是采用过氧化氢为氧化剂时,反应24h后式iii化合物的收率仅有30%左右。
[0041]
所述过氧化物选自过氧化氢、过氧乙酸、叔丁基过氧化氢中的一种或多种;尤其过氧化氢具有价格低、安全性好的优势。过氧化物与2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪的摩尔比为4-10:1,优选6-8:1。
[0042]
本发明中,所述氧化反应在助剂作用下进行,助剂能够促使过氧化物产生更多的羟基自由基进行氧化反应,缩短反应时间,且能使反应更彻底;所述助剂为过渡金属盐、镁盐或氧化物或氢氧化物;所述过渡金属盐优选为铜盐或铁盐,优选地,所述助剂选自氯化镁、硫酸镁、氢氧化镁、氧化镁、氯化亚铁、硫酸铜、氯化铜、碳酸铜中的一种或多种。所述助剂的加入量为5-10mol%,例如可以为6mol%、7mol%、8mol%、9mol%。
[0043]
在氧化反应时,为了保证反应原料和体系的稳定,还可以使用碱调节ph使反应体系为弱碱性,所述碱为固体碱或液体碱。优选地,所述碱为氢氧化钠、氢氧化钾、碳酸氢钠、碳酸氢钾、碳酸钠、碳酸钾及其水溶液中的一种或多种。所述碱的加入以保证体系为弱碱性
为宜,在实际操作中,一般控制ph在7-9范围内。在具体实施例中,还可以选用氢氧化镁、氧化镁等,其既能保持反应体系为碱性,又能缓慢释放镁离子发挥助剂的作用。
[0044]
所述环己基甲醛与2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪的摩尔比为2.0-4.0:1,优选2.1:1。
[0045]
为提高反应收率和减少副产物的产生,控制反应的温度为10-80℃,优选40-65℃,在具体实施方式中,该温度可以为45℃、50℃、55℃或60℃。
[0046]
所述反应还包括采用结晶法提纯式iii所示的目标产物的步骤;所述结晶时溶剂选自石油醚、正庚烷、正辛烷、正戊烷、环己烷中的至少一种。
[0047]
在优选实施方式中,在结晶前进行萃取的步骤,所述萃取剂选自二氯甲烷、环己烷、乙酸乙酯、正庚烷中的至少一种。优选地,在萃取前用还原剂中和过量的过氧化物,或萃取后用还原剂洗涤有机相。所述还原剂为本领域技术人员公知的还原剂即可,比如,所述还原剂选择维生素c。在实际操作中,一般会先通过淀粉碘化钾试纸检测是否有过氧化物剩余,当没有过氧化物剩余或经过一段时间放置已不存在过氧化物时,则对反应液直接萃取和结晶即可。
[0048]
在本发明的另一方面,提供了一种n-烷氧基受阻胺光稳定剂的制备方法,采用上述任一种方法制得式iii所述的n-烷氧基受阻胺光稳定剂中间体,中间体与三乙四胺或硅烷偶联剂kh550反应,制得如下式iv和v的n-烷氧基化的受阻胺光稳定剂:
[0049][0050]
本发明所用原料2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪已有相对比较成熟的制备方法,可以通过已公开的文献中记载的方法制备,例如cn110437207a中公开了通过三聚氯氰与2,2,6,6-四甲基哌啶丁胺反应制得该原料。
[0051]
本发明所用其他过氧化物以及其他试剂均可商购获得,或者采用本领域已知的技术手段制备获得。
[0052]
本发明所述“mol%”为摩尔百分比,即相对于参照物是1mol时,5mol%是指0.05mol;10mol%是指0.10mol。
[0053]
以下实施例用于说明本发明,但不用来限制本发明的范围。实施例中涉及的操作如无特殊说明,均为本领域常规技术操作。
[0054]
以下实施例中的hals中间体制备的反应式为:
[0055][0056]
式i化合物:2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪的制备
[0057]
取500ml三口烧瓶加入100ml间二甲苯作为溶液,18.40g(0.1mol)三聚氯氰,在常温下进行搅拌并缓慢滴加100ml 2,2,6,6,-四甲基哌啶丁胺的间二甲苯溶液(2.1mol/l),大约三十分钟滴加完毕,室温继续搅拌30min,然后加入2.2ml质量分数为30%的氢氧化钠水溶液,缓慢升温60℃,反应10h,反应完毕后,浓缩,并加入大量水稀释,搅拌析出白色固体,过滤。得到50.2g产物,收率93.6%。
[0058]
实施例1 式iii化合物:n-烷氧基受阻胺光稳定剂中间体制备
[0059]
取10.00g(18.65mmol)2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪加入到500ml的三口圆底烧瓶,两侧装配恒压滴液漏斗,中间装配直形冷凝管。向反应容器中加入200mg mgo作为反应助剂。加入100ml间二甲苯作为反应溶剂,使用1%的氢氧化钠水溶液调节ph为7-8,缓慢升温至40℃,同时不断搅拌,缓慢滴加15ml(150mmol)30%过氧化氢水溶液,约8h左右滴加完毕,在滴加过氧化氢水溶液的同时缓慢加入150mg cu(oac)
2
,并滴加4.20g(37.5mmol)环己基甲醛,反应8h。tlc检测反应进程。
[0060]
反应结束后,使用淀粉碘化钾试纸进行过氧化物检测,若淀粉碘化钾试纸变蓝,使用等体积水进行稀释,之后使用二氯甲烷进行萃取,分相,将有机相使用20ml 10%的维生素c水溶液溶液进行洗涤,干燥、浓缩后使用石油醚进行重结晶。得到3.76g白色固体粉末,收率30%,纯度96%。
[0061]
实施例2 式iii化合物:n-烷氧基受阻胺光稳定剂中间体制备
[0062]
取10.00g(18.65mmol)上述方法制得的2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪加入到500ml的三口圆底烧瓶,两侧装配恒压滴液漏斗,中间装配直形冷凝管。向反应容器中加入0.50g mgcl
2
作为反应助剂。加入80ml甲醇作为反应溶剂,使用1%的氢氧化钠水溶液调节ph为7-8,缓慢升温至40℃,同时不断搅拌,缓慢滴加18ml(180mmol)30%过氧化氢水溶液,约8h左右滴加完毕。滴加过氧化氢的同时缓慢加入120mg cucl,并滴加4.17g(37.12mmol)环己基甲醛,继续反应8h。tlc检测反应进程。
[0063]
反应结束后,使用淀粉碘化钾试纸进行过氧化物检测,若淀粉碘化钾试纸变蓝,使用等体积水进行稀释,之后使用二氯甲烷进行萃取,分相,将有机相使用20ml 10%的维生素c水溶液溶液进行洗涤,干燥、浓缩后使用石油醚进行重结晶。得到7.67g白色固体粉末,收率63%,纯度96%。
[0064]
实施例3 式iii化合物:n-烷氧基受阻胺光稳定剂中间体制备
[0065]
取10.00g(18.65mmol)上述方法制得的2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪加入到500ml的三口圆底烧瓶,两侧装配恒压滴液漏斗,中间装配直形冷凝管。向反应容器中加入0.50g mgcl
2
作为氧化反应助剂。加入80ml甲醇作为反应溶剂,使
用1%的氢氧化钠水溶液调节ph为7-8,缓慢升温至40℃,同时不断搅拌,缓慢滴加18ml(180mmol)30%过氧化氢水溶液,在8h左右滴加完毕。滴加过氧化氢的同时缓慢加入180mg cu(oac)
2
,并滴加4.17g(37.12mmol)环己基甲醛,继续反应8h。tlc检测反应进程。
[0066]
反应结束后,使用淀粉碘化钾试纸进行过氧化物检测,如果试纸变蓝则缓慢加入10%的维生素c水溶液除去过氧化物,直至淀粉碘化钾试纸不变蓝,使用等体积水进行稀释,之后使用二氯甲烷进行萃取,分相,将有机相干燥、浓缩后使用正庚烷进行重结晶。得到11.7g白色固体粉末,收率94%,纯度98%。
[0067]
实施例4 式iii化合物:n-烷氧基受阻胺光稳定剂中间体制备
[0068]
取10.00g(18.65mmol)2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪加入到500ml的三口圆底烧瓶,两侧装配恒压滴液漏斗,中间装配直形冷凝管。向反应容器中加入200mg fecl
2
作为反应助剂。加入150ml乙酸乙酯作为反应溶剂,使用1%的氢氧化钠水溶液调节ph为7-8,缓慢升温至50℃,同时不断搅拌,缓慢滴加8ml(80mmol)30%过氧化氢水溶液,约8h左右滴加完毕,在氧化反应进行2h后,缓慢加入170mg cu(oac)
2
,并滴加8.35g(74.2mmol)环己基甲醛,反应8h。tlc检测反应进程。
[0069]
反应结束后,使用淀粉碘化钾试纸进行过氧化物检测,若淀粉碘化钾试纸变蓝,使用等体积水进行稀释,之后使用二氯甲烷进行萃取,分相,将有机相使用20ml 10%的维生素c水溶液溶液进行洗涤,干燥、浓缩后使用石油醚进行重结晶。得到11.31g白色固体粉末,收率93%,纯度98%。
[0070]
实施例5 式iii化合物:n-烷氧基受阻胺光稳定剂中间体制备
[0071]
取10.00g(18.65mmol)上述方法制得的2-氯-4,6-双((2,2,6,6-四甲基哌啶-4-基)丁基氨基)三嗪加入到500ml的三口圆底烧瓶,两侧装配恒压滴液漏斗,中间装配直形冷凝管。加入200ml乙醇作为反应溶剂,使用1%的氢氧化钠水溶液调节ph为7-8,缓慢升温至65℃,同时不断搅拌,缓慢滴加10ml(100mmol)30%过氧化氢水溶液,约8h左右滴加完毕。滴加过氧化氢的同时缓慢加入110mg cucl,并滴加4.39g(39.17mmol)环己基甲醛,反应10h。tlc检测反应进程。
[0072]
反应结束后,使用淀粉碘化钾试纸进行过氧化物检测,如果试纸变蓝则缓慢加入10%的维生素c水溶液除去过氧化物,直至淀粉碘化钾试纸不变蓝,使用等体积水进行稀释,使用二氯甲烷进行萃取,分相,将有机相干燥、浓缩后使用环己烷进行重结晶。得到10.71g白色固体粉末,收率88%,纯度97%。
[0073]
实施例6 n烷氧基受阻胺光稳定剂nor 116的制备
[0074]
取实施例1制备的2g(2.7mmol)1.2-氯-4,6-双[n-(1-环己氧基-2,2,6,6-四甲基哌啶-4-基)丁基氨基]三嗪加入到100ml高压釜,使用20ml间二甲苯作溶剂,加入0.158g(0.9mmol)四胺和2.2ml20%naoh水溶液,n
2
置换三次后冲入2mpan
2
。200℃反应24h。tlc检测(pe:dcm=2:1)反应进行。
[0075]
反应结束后,浓缩反应液,使用甲醇冲洗,得到白色固体1.73g,纯度85%。结构如下:
[0076][0077]
虽然,上文中已经用一般性说明、具体实施方式及试验,对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips