
 商标分类
商标分类  商标转让
商标转让 
基于酶切原理可降低放射性肾浓聚的探针及其制备方法与流程
 2021-02-02 09:02:29|
2021-02-02 09:02:29| 406|
406| 起点商标网
起点商标网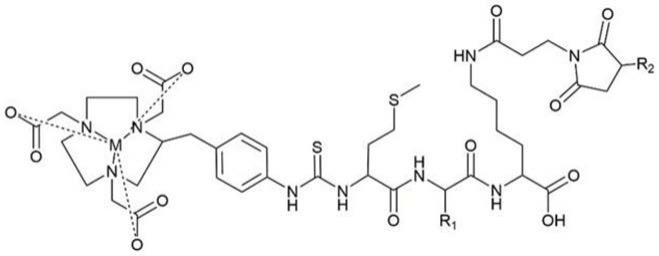
[0001]
本发明涉及医药技术领域,具体涉及一种基于酶切原理可降低放射性肾浓聚的探针及其制备方法。
背景技术:
[0002]
随着分子生物学技术的广泛应用和放射性核素示踪技术的迅速发展,分子核医学在分子功能显像和分子靶向治疗领域取得了长足的发展,使其在精准医疗领域占有重要的一席之地。但是,由于许多肿瘤靶向的配体,如多肽、叶酸、核酸适配体等在体内主要经由肾脏代谢,且代谢产物由于肾的重吸收作用会持续滞留于肾,因而导致与靶向配体连接的放射性核素长时间过高地浓聚于肾。
[0003]
相比于核素显像,核素治疗使用的放射性核素半衰期更长,起始使用剂量更高,因而对肾脏造成的损伤也更严重,详述如下:
[0004]
1、肾脏是放射性辐射的易受损器官。既往辐射生物学研究表明,当肾脏累计剂量超过23gy的吸收剂量后,5年内发展为放射性肾病的概率为5%;而当累计剂量超过28gy时,该概率则跃升至50%。若在5周内肾脏累积照射剂量>20gy,约37%的病人可发生放射性纤维化和少尿性肾衰竭。
[0005]
2、放射性肾损伤的发生率高、后果严重。一项
90
y-dotatoc的治疗研究显示,在未进行任何肾保护措施下,接受4-6个标准治疗疗程的病人中,有28.6%的患者表现为进展性2级肾毒性或因急性肌酐增高而终止治疗,57.1%的患者表现为轻度肾毒性(1级,肌酐1.13
–
1.3mg/dl)。
[0006]
3、放射性肾损伤具有迟发型和隐匿性,难以灵敏监测。辐射生物学研究表明,肾脏在极高剂量照射后,需经6个月至1年潜伏期后方可引起明显的临床症状(如蛋白尿,不同程度的肾功能不全,贫血,低血压);在5周内肾脏多次小剂量照射的累积剂量>20gy,约37%的病人可在6个月后发生放射性纤维化和少尿性肾衰竭。
[0007]
4、放射性肾损伤的剂量阈值不确定,预防难度大。诸多危险因子(如年龄超过60岁、患有糖尿病、高血压、化疗史等)会进一步降低肾对吸收剂量的耐受性达24.3%-30%。
[0008]
综上所述,受体介导的放射性核素治疗中,存在放射性肾损伤发生率高、后果严重、阈值不确定、难以监控和预测的突出问题,因此,肾脏成为了受体介导的放射性核素治疗的主要剂量限定器官,极大地制约了治疗的给药剂量、疗程数和最终的疗效,成为受体介导的放射性核素治疗亟待解决的关键性问题。
技术实现要素:
[0009]
本发明的目的是提供一种基于酶切原理可降低放射性肾浓聚的探针及其制备方法,能有效降低病人在核素诊疗过程中受到的不必要的辐射剂量。
[0010]
本发明所采用的技术方案为:
[0011]
基于酶切原理可降低放射性肾浓聚的探针,其特征在于:
90℃反应5-40min,冷却;加水稀释反应液后经sep-pak c18色谱柱分离纯化,以缓冲液或水冲洗色谱柱以除去未反应的放射性离子,以盐酸乙醇溶液或乙醇溶液淋洗,再经生理盐水或pbs稀释后无菌过滤,即得到放射性标记的配合物的注射液,放射性标记的配合物结构为:
[0035][0036]
其中,r
1
是疏水氨基酸的残基、r
2
是靶向配体、m为放射性核素。
[0037]
冻干法标记法包括以下步骤:
[0038]
将所述探针和其他必要试剂溶于缓冲液中,所得溶液经无菌过滤后分装于冻存管中,经冷冻干燥后密封得到冻干药盒;向冻干药盒中加入缓冲溶液溶解,再加入新鲜制的放射性溶液,密闭37-120℃反应5-40min,冷却;加水稀释反应液后经sep-pak c18色谱柱分离纯化,以缓冲液或水冲洗色谱柱以除去未反应的放射性离子,以盐酸乙醇溶液或乙醇溶液淋洗,再经生理盐水或pbs稀释后无菌过滤得到放射性标记的配合物的注射液,放射性标记的配合物结构为:
[0039][0040]
其中,r
1
是疏水氨基酸的残基,r
2
是靶向配体,m为放射性核素。
[0041]
本发明具有以下优点:
[0042]
与现有技术相比,本发明所述的具有肿瘤靶向的放射性配合物作为核素显像探针或放射性核素治疗探针,应用于人或动物时不仅具有优异的显像或治疗效果,同时可以显著地降低肾脏的放射性浓聚与滞留,具有非常高的靶/非靶比值,极大地降低了应用于人或动物时的辐射剂量,更加安全有效,具有极高的临床推广价值。
附图说明
[0043]
图1为实施例1制备的化合物4-1(a)、4-2(b)、4-3(c)及4-4(d)的hplc分析图及lc-ms图。
[0044]
图2为实施例2制备的化合物5-2(a)、5-3(b)的hplc分析图及lc-ms图。
[0045]
图3为实施例3制备的化合物7-2的hplc分析图及lc-ms图。
[0046]
图4为实施例5制备的化合物8的hplc分析图(放射性检测),及其在pbs及鼠血清中
放置2h的hplc分析图(放射性检测)。图a-d分别为化合物8-1、8-2、8-3及8-4在pbs中的稳定性测定图;图e-h分别为化合物8-1、8-2、8-3及8-4在鼠血清中的稳定性测定图;
[0047]
图5为实施例5制备的化合物8-1(a)、8-2(b)、8-3(c)及8-4(d)在正常小鼠肾脏及尿液中的代谢结果。
[0048]
图6为实施例5制备的化合物8(8-2、8-3及8-4)在荷ins-1瘤裸鼠体内的pet显像图。
[0049]
图7为实施例5制备的化合物8(8-2、8-3及8-4)在荷ins-1瘤裸鼠体内的肿瘤摄取和肾摄取结果。
[0050]
图8为实施例7制备的化合物10(10-2及10-3)在正常鼠体内的肾摄取结果。
[0051]
图9为实施例8制备的化合物11在正常鼠体内的肾摄取结果。
[0052]
图10为实施例9制备的化合物12在荷22rv1瘤裸鼠的micropet显像图。
[0053]
图11为实施例9制备的化合物12在荷22rv1瘤裸鼠体内的肿瘤摄取和肾摄取结果。
[0054]
图12为实施例10制备的化合物13在正常鼠生物分布中肾的量化结果。
具体实施方式
[0055]
下面结合具体实施方式对本发明进行详细的说明。
[0056]
中性内肽酶(neutral endopeptidase,nep)是一种广泛存在于肾刷状缘的肽酶,其可以选择性地从n端剪切疏水性的氨基酸。基于此,我们前期设计并合成了nep的酶解底物甲硫氨酸-缬氨酸-赖氨酸(met-val-lys),并将其插入到双功能螯合剂与her2 affibody之间,借助nep在met与val之间进行的切割,从而释放出核素-螯合剂-met的复合体。由于该复合体不能被肾小管重新收,因而很快到达膀胱,并随尿被排除体内。本发明在上述研究基础上发现,若将多肽序列中的第二个氨基酸替换为疏水性更强或空间位阻更大的氨基酸(如苯丙氨酸phe、色氨酸trp等)时,其酶切效率更快,降低放射性肾浓聚的作用也更强。且本发明经研究发现,该策略不止能降低放射性her2 affibody的肾浓聚,还可以降低放射性标记的exendin 4和psma等配体的肾浓聚,表明该策略对放射性配体具有普适性。
[0057]
综上,本申请提出了一种降低放射性探针肾浓聚的策略,即在放射性核素与多肽之间引入可酶切的三肽序列,该序列的第一个氨基酸为met,第三个氨基酸为lys,第二个氨基酸为具有较大侧链的疏水性氨基酸,且侧链空间位阻越大的疏水性氨基酸,其酶切效率越高,降低放射性肾浓聚的效果越好。该策略有望应用到其他的放射性配体中。该策略有望显著降低病人在核素诊疗过程中受到的不必要的辐射剂量,具有强大的临床应用前景。
[0058]
本发明提供了一类含酶切序列可降低放射性肾浓聚的探针,这里的酶切序列通式为met-x-lys,其中x为疏水性氨基酸;所述的探针结果通式如下式(-)所示,其中,r
1
指双功能螯合剂基团,r
2
指疏水性氨基酸的侧链残基,r
3
指不同的配体分子。
[0059][0060]
本发明所述的方案中,所述的双功能螯合剂可以是现有技术中的各种可用于螯合放射性核素的双功能连接剂;本发明优选的双功能螯合剂是p-scn-bn-nota和dota-nhs-ester;即所述式(i)中的r
1
是结构如式(-)所示的p-scn-bn-nota和结构如式(-)所示的dota-nhs-ester;
[0061][0062]
本发明所述的方案中,所述的疏水性氨基酸包括甘氨酸(gly,g)、丙氨酸(ala,a)、缬氨酸(val,v)、亮氨酸(leu,l)异亮氨酸(ile,i)、苯丙氨酸(phe,f)、脯氨酸(pro,p)、酪氨酸(tyr,y)、色氨酸(trp,w);本发明优选的疏水性氨基酸为phe、tyr、trp,即所述式(i)中的r
2
是结构如式(-)所示的phe、结构如式(
ⅴ
)所示的trp或结构如式(
ⅵ
)所示的trp;
[0063]
本发明所述的方案中,所述的配体可以是现有技术中的各种肿瘤靶向的配体
[0064][0065]
分子;本发明优选的配体分子是exendin 4、psma-617、her2亲合体,即所述式(i)中的r
3
是结构如式(
ⅶ
)所示的exendin 4、结构如式(
ⅷ
)所示的psma-617或、结构如式(
ⅸ
)所示的her2亲合体、结构如(
ⅹ
)所示的奥曲肽类似物或结构如(
ⅺ
)所示的叶酸;
[0066][0067]
式(
ⅶ
)所示的exendin 4序列为gegtftsdlskqleeeavrlfiewlknggpssgapppsc-nh
2
;
[0068][0069]
her2亲合体是结构如式(
ⅸ
)所示的靶向分子z
her2:2891
,其序列为nh
2-aeakyakemrnayweiallpnltnqqkrafirklyddpsqssellseakklndsqapkc-cooh;
[0070][0071]
本发明所述的化合物结构中含有用于肿瘤靶向的配体分子、可被肾刷状缘nep酶解的三肽序列mxk,以及用于鳌合放射性核素的双功能连接剂,可在保持肿瘤摄取不变的情况下,显著降低肾中的放射性浓聚与滞留,极大地提升了放射性配体的临床诊疗价值,具有明确的临床应用前景。
[0072]
本发明还提供一种制备所述化合物的方法,合成路线如下:
[0073][0074]
如上述合成路线所示,所述制备方法具体包括以下步骤:
[0075]
1)将保护的赖氨酸与2-氯三苯基氯树脂以1.2:1的摩尔比混合,在n,n-二异丙基乙胺(dipea)作用下,将保护的赖氨酸连接到树脂上;
[0076]
2)在步骤1)所得产物的基础上,继续连接氨基酸x和boc-met,得到第二步产物;
[0077]
3)步骤2)得到的第二步产物在2%的水合肼的作用下,脱去保护剂dde,得到第三步产物;
[0078]
4)步骤3)得到的第三步产物在20%的1,1,1,3,3,3-六氟-2-丙醇(hfip)的作用下从树脂上切割下来,得到第四步产物;
[0079]
5)步骤4)得到的第四步产物在三乙胺的作用下,与3-(马来酰亚胺基)丙酸n-羟基琥珀酰亚胺酯反应,得到第五步反应产物;
[0080]
6)步骤5)得到的第五步产物在三氟乙酸(tfa)条件下脱去保护基团boc,得到第六步反应产物;
[0081]
7)步骤6)得到的第六步产物在dipea作用下,与双功能螯合剂反应,得到第七步产物;
[0082]
8)步骤7)得到的第七步产物在pbs中与肿瘤靶向配体反应,得到本发明所述的配体化合物。
[0083]
本发明所述的方案中,步骤2)所述的氨基酸x选自疏水性氨基酸gly、ala、val、leu、ile、phe、pro、tyr、trp;本发明优选的疏水性氨基酸为phe、tyr、trp。
[0084]
本发明所述的方案中,步骤7)所述的双功能螯合剂可以选自p-scn-bn-nota、
nota-nhs-ester、p-scn-bn-dota、dota-nhs-ester、p-ncs-bz-dfo或p-scn-bn-dtpa;优选p-scn-bn-nota和dota-nhs-ester。
[0085]
本发明所述的方案中,步骤8)所述的肿瘤靶向配体exendin 4可以选自cys
40-exedin 4、cys
40-leu
14-exedin 4,优选cys
40-leu
14-exedin 4;psma-617可以选自thiol-psma-617;her2亲合体可以选自z
her2:2891
、z
her2:2395
或z
her2:342
,优选z
her2:2891
;奥曲肽类似物可以选自thiol-tate、thiol-noc、thiol-toc,优选thiol-tate;叶酸类化合物可以选自thiol-folate acid。
[0086]
在此基础上,本发明进一步提供此类化合物的放射性标记配合物,它由本发明所述的化合物经放射性核素标记得到。所述的放射性标记配合物可以作为新型的肿瘤放射性诊疗探针,即可以作为放射性核素诊断探针或放射性核素治疗探针。
[0087]
本发明所述的放射性标记配合物中,所述的放射性诊断核素可以选择核素
18
f、
68
ga、
64
cu、
62
cu、
86
y或
89
zr中的任意一种,优选
18
f、
68
ga、
64
cu中的任意一种;所述的放射性治疗核素可以选择
111
in、
177
lu、
90
y、
225
ac或
213
bi中的任意一种,优选
177
lu、
90
y中的任意一种。
[0088]
本发明所述的放射性标记配合物可以通过含放射性核素的化合物与本发明所述的化合物按照现有的多种标记方法制备得到;本发明优选的标记方法为下述的湿法或冻干法:
[0089]
湿法标记方案,包括:将适量本发明所述的化合物溶于缓冲溶液或去离子水中;在所得溶液中加入放射性核素溶液,密闭反应5-40min,即生成放射性核素标记的配合物;
[0090]
或者,冻干法标记方案,包括:将适量本发明所述的化合物溶于缓冲溶液或去离子水中;将所得溶液经无菌过滤后,分装于容器中,经冷冻干燥后加塞密封,得到冻干药盒;向所述冻干药盒中加入适量乙酸溶液或缓冲液溶解,再加入相应的放射性核素溶液,密闭反应5-40min,即生成放射性核素标记的配合物。其中,所述的分装用容器优选为冻存管或管制抗生素瓶。还可以根据药盒冻干粉成型情况选择在药盒中增加赋形剂,比如甘露醇、抗坏血酸等,并通过调节本发明所述的化合物及赋形剂的用量,使药盒成型达到最佳。
[0091]
所述的湿法标记方案和冻干标记方案得到的产物均可经常规处理(如经色谱分离纯化、旋蒸除去溶剂、以pbs或水或生理盐水溶解剩余物、无菌过滤等)进一步制成注射液。
[0092]
本发明一种优选的所述放射性标记配合物制备方法,是放射性核素的湿法标记法,包括:将本发明所述的化合物4溶于缓冲溶液或去离子水中;在其中加入新鲜淋洗得到的放射性溶液,密闭37℃(nota的化合物)或90℃(dota的化合物)反应5-40min,冷却;加水稀释反应液后经sep-pak c18色谱柱分离纯化,以缓冲液或水冲洗色谱柱以除去未反应的放射性离子,以盐酸乙醇溶液或乙醇溶液淋洗,再经生理盐水或pbs稀释后无菌过滤,即得到结构如式(
ⅻ
)所述的放射性标记的配合物的注射液。其中r
2
是phe、tyr或trp中的一种,r
3
是cys
40-leu
14-exedin 4、thiol-psma-617、z
her2:2891
、thiol-tate或thiol-folate acid中的一种。
[0093]
本发明另一种优选的所述放射性标记配合物制备方法,是冻干法标记法,包括:将本发明所述的化合物和氯化铝溶于缓冲液中,所得溶液经无菌过滤后分装于冻存管中,经冷冻干燥后密封得到冻干药盒;向冻干药盒中加入适量乙酸溶液或缓冲溶液溶解,再加入乙腈或乙醇和新鲜制得的放射性溶液,密闭70-120℃反应5-30min,冷却;加水稀释反应液后经sep-pak c18色谱柱分离纯化,以缓冲液或水冲洗色谱柱以除去未反应的放射性离子,
以盐酸乙醇溶液或乙醇溶液淋洗,再经生理盐水或pbs稀释后无菌过滤得到结构如下式(
ⅻ
)所示的放射性标记的配合物的注射液。其中r
2
是phe、tyr或trp中的一种,r
3
是cys
40-leu
14-exedin 4、thiol-psma-617、z
her2:2891
、thiol-tate或thiol-folate acid中的一种。
[0094][0095]
上述方法中,所述的缓冲溶液为稳定反应液ph的物质,可以为醋酸盐、乳酸盐、酒石酸盐、苹果酸盐、马来酸盐、琥珀酸盐、抗坏血酸盐、碳酸盐或磷酸盐中的任意一种或两种以上的混合物。
[0096]
本发明还提供所述的放射性标记配合物在制备用于人或动物的肿瘤靶向放射性诊疗探针中的应用。
[0097]
与现有技术相比,本发明所述的具有肿瘤靶向的放射性配合物作为核素显像探针或放射性核素治疗探针,应用于人或动物时不仅具有优异的显像或治疗效果,同时可以显著地降低肾脏的放射性浓聚与滞留,具有非常高的靶/非靶比值,极大地降低了应用于人或动物时的辐射剂量,更加安全有效,具有极高的临床推广价值。
[0098]
以下通过具体的实施例和应用例来进一步说明本发明:其中合成步骤中所使用的化学物质均为现有物质实施例或市售商品。
[0099]
实施例1
[0100]
制备一类含有酶切序列的exendin 4化合物,其合成路线如下:
gly-oh、fmoc-val-oh、fmoc-phe-oh或fmoc-trp-oh,4当量)和boc-met-oh(0.399g,4当量)以增长肽链。固相合成结束后,将连有树脂的多肽粗产品置于3ml 2%水合肼的dmf溶液中,室温下搅拌1.5h,抽滤,依次用二氯甲烷、甲醇和dmf洗涤树脂。之后将该树脂置于3ml20%hfip的二氯甲烷溶液中,室温下搅拌2h,抽滤,取滤液,经乙醚重结晶后,即得到化合物1,产率约为70-84%。
[0106]
称取0.08mmol的化合物1于10ml玻璃瓶中,依次加入1.2当量的3-(马来酰亚胺基)丙酸n-羟基琥珀酰亚胺酯,2.6当量的三乙胺,500μl dmso,于室温下搅拌反应。用hplc监测反应进度,当观察到原料消失时,加入500μl 0.1%tfa水溶液终止反应。用制备型hplc对产物进行分离纯化,得化合物2,产率约为45-56%。经lc-ms鉴定,化合物2-1至2-4的分子量分别为586[m+h]
+
,626[m-h]-,674[m-h]-和815[m+h]
+
(trp侧链的胺基被boc保护)。
[0107]
2、化合物3的合成:
[0108]
称取0.03mmol的化合物2置于10ml玻璃瓶中,加入500μl tfa,室温下静置20min,用hplc检测反应进度,当所有原料都转化为产物时,停止反应。用氮气将tfa吹干。向反应瓶中加入500μl dmso,1当量的nota-ncs和5当量的三乙胺,于室温下搅拌反应。用hplc检测反应进度,当观察到原料消失时,加入500μl 0.1%tfa水溶液终止反应。用制备型hplc对产物进行分离纯化,得化合物3,产率约为46-50%。经lc-ms鉴定,化合物3-1至3-4的分子量分别为934[m-h]-,976[m-h]-,1024[m-h]-和1064[m-h]-。
[0109]
3、化合物4的合成:
[0110]
将化合物3(0.66μmol,1.5当量)加入到cys
40-leu
14-exedin 4(市售,0.44μmol,1当量)的pbs溶液中,室温下反应两小时后用hplc分离纯化,得化合物4,产率约为50-59%。经lc-ms鉴定,化合物4-1至4-4的分子量分别为5264[m-h]-(钴或镍的螯合物),5268[m+h]
+
(钠的螯合物),5297[m+h]
+
and 5337[m+h]
+
。化合物4-1至4-4的hplc分析图及lc-ms图见图1所示。
[0111]
实施例2
[0112]
制备一类含有酶切序列的z
her2:2891
的化合物,其合成路线如下:
[0113][0114]
具体制备过程包括以下步骤:
[0115]
1、化合物1-2、1-3、2-2、2-3、3-2及3-3的合成同实施例一;
[0116]
2、化合物5的合成:
[0117]
将化合物3(0.66μmol,1.5当量)加入到z
her2:2891
(市售,3mg,0.44μmol,1当量)的pbs溶液中,室温下反应两小时后用hplc分离纯化,得化合物5,产率约为54-62%。经lc-ms鉴定,化合物5-2及5-3的分子量分别为[m+h]
+
=7794.00和[m+7h]
7+
=1121.50,计算值(m/z)为7793.90(c
343
h
543
n
93
o
106
s
4
)和7841.90(c
347
h
543
n
93
o
106
s
4
)。化合物5-2及5-3的hplc分析图及lc-ms图见图2所示。
[0118]
实施例3
[0119]
制备一类含有酶切序列的psma的化合物,其合成路线如下:
[0120][0121]
具体制备过程包括以下步骤:
[0122]
1、化合物1-2及2-2的合成同实施例一;
[0123]
2、化合物6-2的合成:
[0124]
称取0.03mmol的化合物2-2置于10ml玻璃瓶中,加入500μl tfa,室温下静置20min,用hplc检测反应进度,当所有原料都转化为产物时,停止反应。用氮气将tfa吹干。向反应瓶中加入500μl dmso,1当量的dota-nhs-ester和5当量的三乙胺,于室温下搅拌反应。用hplc检测反应进度,当观察到原料消失时,加入500μl 0.1%tfa水溶液终止反应。用制备型hplc对产物进行分离纯化,得化合物5,产率约为40-48%。经lc-ms鉴定,化合物6-2的分子量为913[m-h]-,计算值(m/z)为914(c
39
h
63
n
9
o
14
s)。
[0125]
3、化合物7-2的合成:
[0126]
将化合物6-2(6.6μmol,1.5当量)加入到thiol-psma-617(市售,3.4mg,4.4μmol,1当量)的pbs溶液中,室温下反应两小时后用hplc分离纯化,得化合物6,产率约为46-52%。经lc-ms鉴定,化合物7-2的分子量为1644.7[m+h]
+
,计算值分别(m/z)为1643.9(c
74
h
110
n
14
o
24
s
2
)。化合物7-2的hplc分析图见图3所示。
[0127]
实施例4.放射性标记用冻干药盒的制备
[0128]
1)放射性ga-68和cu-64标记用冻干药盒的制备(以制备100支为例)
[0129]
称取4mg实施例1制备的化合物4溶于40ml 0.5mol/l的醋酸-醋酸钠缓冲溶液(ph=4)中,经无菌过滤后分装于400支冻存管中,然后置于冷冻干燥剂中冷冻干燥24小时,加塞密封,得到冻干药盒-。根据药盒冻干粉针成型情况,可在药盒中增加赋形剂,比如甘露醇、抗坏血酸等,可调节化合物4及赋形剂的用量,以使药盒成型达到最佳。
[0130]
2)放射性f-18标记用冻干药盒的制备(以制备100支为例)
[0131]
称取4mg实施例1制备的化合物4溶于10ml 0.5mol/l的酒石酸-酒石酸钾钠缓冲溶液(ph=4)中,再将0.04mg氯化铝(alcl
3
)溶于10ml 0.5mol/l的酒石酸-酒石酸钾钠缓冲溶液(ph=4)中,将二者混合均匀。经无菌过滤后分装于100支冻存管中,然后置于冷冻干燥剂中冷冻干燥24小时,加塞密封,得到冻干药盒-。根据药盒产量以及对每支药盒中组分含量要求的不同,可调节化合物4及氯化铝的用量,使它们的重量比落在(20-100):1范围内。
[0132]
实施例5.一种ga-68标记的放射性诊断探针(化合物8)的制备:
[0133]
1)湿法:将约18.5-1850兆贝可(mbq)
68
gacl
3
盐酸溶液(淋洗自锗镓发生器)加入到含0.5ml实施例1制备的化合物4的醋酸-醋酸盐溶液(1.0g/l)的离心管中,置于37℃下反应20min。取一c18分离小柱,先用10ml无水乙醇缓慢淋洗,再用10ml水淋洗。用10ml水将标记液稀释后,上样到分离柱上,先用10ml水除去未标记的
68
ga离子,再用0.3ml 10mm hcl的乙醇溶液淋洗得到化合物7。该淋洗液经生理盐水稀释,并经无菌过滤后即得化合物8注射液。
[0134]
2)冻干法:将约18.5-1850兆贝可(mbq)
68
gacl
3
盐酸溶液(淋洗自锗镓发生器)加入到含有化合物4的冻干药盒-中,混匀后37℃下反应20min。取一c18分离小柱,先用10ml无水乙醇缓慢淋洗,再用10ml水淋洗。用10ml水将标记液稀释后,上样到分离柱上,先用10ml水除去未标记的
68
ga离子,再用0.3ml10mm hcl的乙醇溶液淋洗得到化合物8。该淋洗液经生理盐水稀释,并经无菌过滤后即得化合物8注射液。
[0135]
实施例6.一种f-18标记的放射性诊断探针(化合物9)的制备:
[0136]
1)湿法:在1ml离心管中,加入3μl 2mm的醋酸-醋酸盐溶液(0.5mol/l,ph=4)和6μl 3mm实施例1制备的化合物4的醋酸-醋酸盐溶液(0.5mol/l,ph=4),然后加入0.13ml的乙腈和0.05ml约37mbq的
18
f-水溶液,充分混合后置于沸水浴中反应10min。取一c18分离小柱,先用10ml无水乙醇缓慢淋洗,再用10ml水淋洗。用10ml水稀释已冷却的标记液,并将其转移至分离柱上,先用10ml水除去未标记的
18
f离子,再用0.3ml 10mmhcl溶液淋洗得到化合物9。该淋洗液经生理盐水稀释,并经无菌过滤后即得化合物8注射液。
[0137]
2)冻干法:向含有化合物4的冻干药盒-中加入0.5ml 0.5mol/l的醋酸-醋酸盐溶液(ph=4),全部溶解后加入约37-3700mbq
18
f-乙腈淋洗液(从阴离子捕获柱qma获得),密闭120℃反应5min,冷却。取一c18分离小柱,先用10ml无水乙醇缓慢淋洗,再用10ml水淋洗。用10ml水稀释已冷却的标记液,并将其转移至分离柱上,先用10ml水除去未标记的
18
f离子,再用0.3ml 10mmhcl溶液淋洗得到化合物9。该淋洗液经生理盐水稀释,并经无菌过滤后即得化合物9注射液。
[0138]
实施例7.一种cu-64标记的放射性诊断探针(化合物10)的制备:
[0139]
1)湿法:将约18.5-1850mbq
64
cucl
2
醋酸钠溶液加入到含0.5ml实施例1制备的化合物4的醋酸-醋酸盐溶液(4.0g/l)的离心管中,置于37℃下反应20min。取一c18分离小柱,先用10ml无水乙醇缓慢淋洗,再用10ml水淋洗。用10ml水将标记液稀释后,上样到分离柱上,先用10ml水除去未标记的
64
cu离子,再用0.3ml 10mmhcl的乙醇溶液淋洗得到化合物10。该淋洗液经生理盐水稀释,并经无菌过滤后即得化合物10注射液。
[0140]
2)冻干法:将约18.5-1850mbq
64
cucl
2
醋酸钠溶液加入到含有化合物4的冻干药盒-中,混匀后37℃下反应20min。取一c18分离小柱,先用10ml无水乙醇缓慢淋洗,再用10ml水淋洗。用10ml水将标记液稀释后,上样到分离柱上,先用10ml水除去未标记的
64
cu离子,再用0.3ml 10mm hcl的乙醇溶液淋洗得到化合物10。该淋洗液经生理盐水稀释,并经无菌过滤
后即得化合物10注射液。
[0141]
实施例8.一种ga-68标记的放射性诊断探针(化合物11)的制备:
[0142]
1)湿法:将约18.5-1850mbq
68
gacl
3
盐酸溶液(淋洗自锗镓发生器)加入到含0.5ml实施例2制备的化合物5的醋酸-醋酸盐溶液(1.0g/l)的离心管中,置于37℃下反应20min。取一c18分离小柱,先用10ml无水乙醇缓慢淋洗,再用10ml水淋洗。用10ml水将标记液稀释后,上样到分离柱上,先用10ml水除去未标记的
68
ga离子,再用0.3ml 10mm hcl的乙醇溶液淋洗得到化合物11。该淋洗液经生理盐水稀释,并经无菌过滤后即得化合物11注射液。
[0143]
2)冻干法:将约18.5-1850mbq
68
gacl
3
盐酸溶液(淋洗自锗镓发生器)加入到含有化合物5的冻干药盒-中,混匀后37℃下反应20min。取一c18分离小柱,先用10ml无水乙醇缓慢淋洗,再用10ml水淋洗。用10ml水将标记液稀释后,上样到分离柱上,先用10ml水除去未标记的
68
ga离子,再用0.3ml 10mm hcl的乙醇溶液淋洗得到化合物11。该淋洗液经生理盐水稀释,并经无菌过滤后即得化合物11注射液。
[0144]
实施例9.一种ga-68标记的放射性诊断探针(化合物12)的制备:
[0145]
1)湿法:将约18.5-1850mbq
68
gacl
3
盐酸溶液(淋洗自锗镓发生器)加入到含0.5ml实施例3制备的化合物7的醋酸-醋酸盐溶液(1.0g/l)的离心管中,置于90℃下反应20min。取一c18分离小柱,先用10ml无水乙醇缓慢淋洗,再用10ml水淋洗。用10ml水将标记液稀释后,上样到分离柱上,先用10ml水除去未标记的
68
ga离子,再用0.3ml 10mm hcl的乙醇溶液淋洗得到化合物12。该淋洗液经生理盐水稀释,并经无菌过滤后即得化合物12注射液。
[0146]
2)冻干法:将约18.5-1850mbq
68
gacl
3
盐酸溶液(淋洗自锗镓发生器)加入到含有化合物7的冻干药盒-中,混匀后90℃下反应20min。取一c18分离小柱,先用10ml无水乙醇缓慢淋洗,再用10ml水淋洗。用10ml水将标记液稀释后,上样到分离柱上,先用10ml水除去未标记的
68
ga离子,再用0.3ml 10mm hcl的乙醇溶液淋洗得到化合物12。该淋洗液经生理盐水稀释,并经无菌过滤后即得化合物12注射液。
[0147]
实施例10.一种lu-177标记的放射性诊断探针(化合物13)的制备:
[0148]
1)湿法:将约18.5-1850mbq 177
lucl
3
醋酸钠溶液加入到含0.5ml实施例3制备的化合物7的醋酸-醋酸盐溶液(1.0g/l)的离心管中,置于90℃下反应20min。取一c18分离小柱,先用10ml无水乙醇缓慢淋洗,再用10ml水淋洗。用10ml水将标记液稀释后,上样到分离柱上,先用10ml水除去未标记的
177
lu离子,再用0.3ml 10mm hcl的乙醇溶液淋洗得到化合物13。该淋洗液经生理盐水稀释,并经无菌过滤后即得化合物13注射液。
[0149]
2)冻干法:将约18.5-1850mbq
64
cucl
2
醋酸钠溶液加入到含有化合物7的冻干药盒-中,混匀后90℃下反应20min。取一c18分离小柱,先用10ml无水乙醇缓慢淋洗,再用10ml水淋洗。用10ml水将标记液稀释后,上样到分离柱上,先用10ml水除去未标记的
177
lu离子,再用0.3ml 10mm hcl的乙醇溶液淋洗得到化合物13。该淋洗液经生理盐水稀释,并经无菌过滤后即得化合物13注射液。
[0150]
实施例11.分析及应用效果
[0151]
以下以实施例1制备的化合物4和实施例5制备的放射性
68
ga标记探针(化合物8)为例,其性能测定描述如下:
[0152]
1、hplc分析鉴定
[0153]
hplc体系如下:agilent 1100;c18色谱柱(zorbax,5μm,4.6
×
250mm)用于多肽分
析。化合物4-1至4-4的保留时间分别16.6、15.4、17.3和15.9min,并以此计算化学纯度大于95%。hplc结果见图1。
[0154]
淋洗梯度:0-3分钟:15%乙腈(0.1%tfa)和85%水(0.1%tfa)保持不变;3-20分钟:增加到70%乙腈(0.1%tfa)和30%水(0.1%tfa)。
[0155]
2、化合物8在pbs及鼠血清中的稳定性分析
[0156]
1)取20μl(7.4mbq)化合物8加入到200μl的pbs溶液中,并置于室温下孵育,分别于1h及2h后用微量注射器取50μl孵育混合物,用radio-hplc测定放射化学纯度。hplc结果见图4。
[0157]
2)取20μl(7.4mbq)化合物8加入到200μl新鲜鼠血清溶液中,并置于37℃孵育,分别于孵育1h及2h后,用微量注射器取50μl孵育混合物,加入等体积的乙腈沉淀蛋白,之后用高速离心机在12000rpm下离心5min,小心取得上清液,用radio-hplc测定放射化学纯度。hplc结果见图4。
[0158]
3.化合物8在正常小鼠肾和尿中的代谢试验
[0159]
按实施例5制备好放射化学纯度大于95%的化合物8溶液,取100μl(约18.5mbq,比活度为389-486mbq/μmol)通过尾静脉注射于正常小鼠。注射探针20min后,通过挤压膀胱收集尿液样本。之后迅速处死小鼠,取出肾脏,置于冰上。将尿液样本与等体积的乙腈混合,置于4℃离心机内,于14,000rpm下离心10min。在新鲜的肾脏组织中加入1ml预冷的酸性缓冲液(0.2m甘氨酸-0.15m nacl,ph 3.0),于冰上高速匀浆90s,之后加入等体积的乙腈沉降蛋白,于4℃离心机内,14,000rpm下离心30min。分别取10μl尿液和肾脏的上清液样本进行radio-hplc分析。radio-hplc的分析条件为:流动相a为含0.1%三氟乙酸的去离子水,流动相b为含0.1%三氟乙酸的乙腈;采用梯度洗脱:0-5min,5-5%b相;5-35min:5-65%b相;流速为1ml/min。
[0160]
实验结果如图5所示,酶切片段ga-nota-met-oh标准品的保留时间为16.2min,8-1至8-4的标准品的保留时间分别为30.2、27.8、28.3和31.5min。注射放射性探针20min后,其在肾脏样本中均有残留,而在尿液样本中则均无相应的原型化合物。
[0161]
化和去8-1的尿液代谢物中,检测到酶切片段ga-nota-met-oh的含量非常少,提示mgk的酶切效率较低;而在化合物8-2至8-4的尿液代谢物中,均检测到了酶切片段ga-nota-met-oh的峰,其占比分别为46%、56%和60%,提示三者的酶切效率是依次增强的。由于化合物7-1的酶切效率较低,故在后续的pet显像中予以排除。
[0162]
4.化合物8在荷ins-1瘤裸鼠的micropet显像试验
[0163]
按实施例5制备好放射化学纯度大于95%的化合物8溶液,取100μl(约3.7mbq)通过尾静脉注射于荷ins-1瘤裸鼠,并于给药后5、20、40、60、120及180min进行pet图像采集。显像结果如图6和图7所示,与未引入酶切序列的对照探针相比,添加有酶切序列的实验组探针肿瘤摄取水平,在各个时间点都没有显著变化;而实验组探针的肾摄取自30min后显著低于对照组探针,且随着酶切序列中第二个氨基酸侧链疏水性的增强及空间位阻的增大,其降低肾摄取的能力越强,即酶切效率方面,mwk>mfk>mvk。
[0164]
实施例12.分析及应用效果
[0165]
以下以实施例1制备的化合物4和实施例7制备的放射性
64
cu标记探针(化合物10)为例,其性能测定描述如下:
[0166]
1、化合物10在正常鼠的micropet显像试验
[0167]
按实施例7制备好放射化学纯度大于95%的化合物10溶液,取100μl(约3.7mbq)通过尾静脉注射于正常鼠,并于给药后1、4、24及48h进行pet图像采集。显像结果如图8所示,在注射探针1h及4h后,与未引入酶切序列的对照探针相比,实验组探针10-2(
64
cu-nota-mvk-psma)和10-3(
64
cu-nota-mfk-psma)可以显著降低放射性在肾中的浓聚,而在注射探针24h和48h后,实验组探针10-2的肾浓聚与对照组探针无明显差异,但是实验组探针10-3的肾浓聚仍显著低于对照组探针。在图8中的任意时刻,实验组探针10-3的肾浓聚都显著低于实验组探针10-2的肾浓聚,再次证明随着酶切序列中第二个氨基酸侧链疏水性的增强及空间位阻的增大,其降低肾摄取的能力越强,即酶切效率方面,mfk>mvk。
[0168]
实施例13.分析及应用效果
[0169]
以下以实施例2制备的化合物5和实施例8制备的放射性
68
ga标记探针(化合物11)为例,其性能测定描述如下:
[0170]
1、化合物11在正常鼠的micropet显像试验
[0171]
按实施例8制备好放射化学纯度大于95%的化合物11溶液,取100μl(约3.7mbq)通过尾静脉注射于正常鼠,并于给药后5、20、40、60、120及180min进行pet图像采集。显像结果如图9所示,对照探针的肾摄取在5min时为38%id/g,之后随时间逐渐增高,肾摄取值在1、2及3h时分别为74%、76%及85%id/g。相较于对照探针,实验探针11-2和11-3在5min时的肾摄取水平与对照探针相当,分别为36%和35%id/g。但是,在随后的时间里,对照组探针的肾摄取明显低于对照组探针,实验探针11-2在1、2及3h的肾摄取分别为42%、21%及16%id/g,分别比对照探针下降了42%、72%和81%;而另一实验探针11-3的肾摄取,比对照探针的肾摄取分别下降了53%、78%和85%,说明两个实验探针均能有效降低affibody的放射性肾摄取。同时,对比两个实验探针,在同一时间点11-3的肾摄取比11-2的更低,说明随着酶切序列中第二个氨基酸侧链疏水性的增强及空间位阻的增大,其降低肾摄取的能力越强,即mfk的酶切效率大于mvk的酶切效率。
[0172]
实施例14.分析及应用效果
[0173]
以下以实施例3制备的化合物7和实施例9制备的放射性
68
ga标记探针(化合物12)为例,其性能测定描述如下:
[0174]
1、化合物12在荷22rv1瘤裸鼠的micropet显像试验
[0175]
按实施例9制备好放射化学纯度大于95%的化合物12溶液,取100μl(约3.7mbq)通过尾静脉注射于荷22rv1瘤裸鼠,并于给药后5、20、40、60、120及180min进行pet图像采集。显像结果如图10和图11所示,与未引入酶切序列的对照探针相比,添加有酶切序列mvk的实验组探针肿瘤摄取水平,在各个时间点都没有显著变化;而实验组探针的肾摄取在各个时间点均显著低于对照组探针,其摄取值约为对照组探针的一半,表明酶切清除策略也适用于psma。
[0176]
实施例15.分析及应用效果
[0177]
以下以实施例3制备的化合物7和实施例10制备的放射性
177
lu标记探针(化合物13)为例,其性能测定描述如下:
[0178]
1、化合物13在正常鼠中的生物分布实验
[0179]
按实施例10制备好放射化学纯度大于95%的化合物13溶液,取100μl(约3.7mbq)
通过尾静脉注射于正常,并于给药后0.5、1、2、4、8及24h将老鼠处死,取感兴趣脏器,称重并测放射性计数。放射性在脏器中的摄取水平以%id/g表示。显像结果如图12所示,与未引入酶切序列的对照探针相比,添加有酶切序列mvk的实验组探针,在注射药物0.5、1、2及4h后,其放射性肾浓聚均显著低于对照组探针。
[0180]
根据前文所述,添加酶切序列后并不改变肿瘤的放射性摄取。同时本实验证实,酶切清除策略可以在起始时刻就起效。鉴于核素治疗起始剂量大,起始肾浓聚高,该酶切策略的应用,无疑在保证肿瘤摄取的前提下,于起始时刻就显著降低了肾浓聚,不但确保了治疗效果,还大大提高了治疗的安全性。在此基础上,由于肾脏的浓聚降低了,我们还可以根据肾脏的耐受剂量,增大起始的治疗剂量或增加治疗的周期数,从而获得更好的治疗效果。
[0181]
综上所述,酶切清除策略可以显著降低放射性探针的肾浓聚,因而在受体介导的核素诊断与治疗中有极强的应用价值与前景。
[0182]
本发明的内容不限于实施例所列举,本领域普通技术人员通过阅读本发明说明书而对本发明技术方案采取的任何等效的变换,均为本发明的权利要求所涵盖。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips